Methods
The is-pero web server is an advanced, automated platform for the end-to-end prediction
and classification of peroxisomal proteins, designed to streamline large-scale proteomics analyses.
By integrating cutting-edge protein language models with specialized machine learning techniques, is-pero
delivers high-accuracy predictions for peroxisomal protein localisation directly from raw FASTA sequences
or UniProt identifiers.
Developed through a collaboration between the University of Bologna, Wageningen University,
and Ruhr University Bochum, is-pero simplifies peroxisomal protein identification using a
structured, four-module workflow that addresses the extreme class imbalance (1.25% peroxisomal vs. 98.75% non-peroxisomal
proteins) through Synthetic Minority Over-sampling Technique (SMOTE) and cluster-stratified cross-validation.
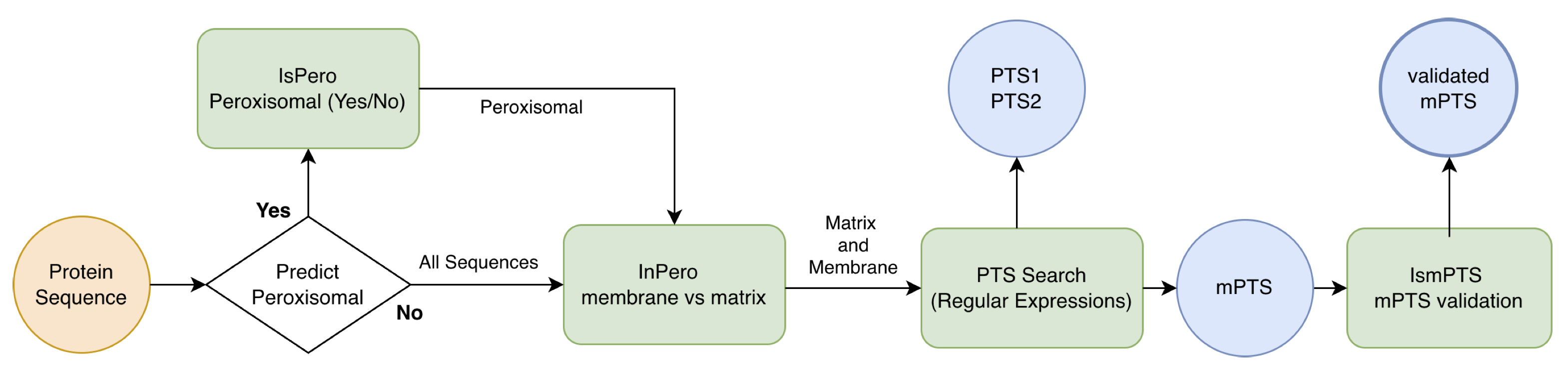
Key Workflow Steps
The is-pero web server integrates four specialized modules that operate sequentially, with the first module serving as an optional initial filtering step. This flexible architecture allows users to either perform complete end-to-end analysis starting from raw sequences, or to directly analyze pre-identified peroxisomal proteins by bypassing the initial classification stage.
Module 1 (is-pero) - Optional Binary Classification: This initial module performs binary classification powered by logistic regression with L2 regularization to distinguish peroxisomal from non-peroxisomal proteins. By leveraging ESM-2 embeddings, it transforms protein sequences into comprehensive numerical representations for robust classification. Users can optionally bypass this module when analyzing already-confirmed peroxisomal proteins.
Module 2 (in-pero): For proteins identified as peroxisomal (either through Module 1 or direct user input), an advanced multi-layer perceptron (MLP) architecture classifies them as either matrix or membrane-associated. This module provides detailed sub-compartment discrimination with probability scores indicating confidence in localisation assignments.
Module 3 (PTS-scan): This deterministic identification module detects canonical peroxisomal targeting signals including PTS1 (C-terminal tripeptide motifs), PTS2 (N-terminal nonapeptide motifs), and membrane peroxisomal targeting signals (mPTS). By combining position-specific scoring matrices with regular-expression pattern matching, it provides transparent reporting of matched peptide sequences and their positions within proteins.
Module 4 (is-mPTS): A specialized logistic regression model predicts PEX19-binding sites in membrane-associated proteins. This module extracts local sequence window representations from full-sequence ESM-2 embeddings to identify membrane-targeting signals that may not be captured by traditional motif-based approaches, enabling discovery of alternative peroxisomal targeting mechanisms.
All modules are powered by ESM-2 protein language model embeddings, with specialized strategies for processing proteins of varying lengths. The integrated workflow generates comprehensive outputs including localisation probabilities, sub-compartment assignments, identified targeting signals with confidence scores, and predicted membrane-targeting regions, available through both interactive web interfaces and standardized CSV formats for seamless integration into proteomics research pipelines. The flow chart of the method is represented in the figure below.
Datasets
The performance of is-pero was rigorously benchmarked using multiple complementary datasets designed to evaluate different aspects of peroxisomal protein prediction:
Binary Classification Benchmark: The core binary classifier was evaluated on a large-scale dataset comprising 135,435 proteins from SwissProt, including 1,674 experimentally validated peroxisomal proteins and 133,761 non-peroxisomal proteins. This dataset reflects the extreme class imbalance typical of organellar localisation tasks (<1.25% positive class).
Sub-compartment Discrimination Dataset: For sub-localisation prediction, a curated dataset of 304 peroxisomal proteins was used, including both matrix and membrane proteins with experimentally validated annotations, allowing evaluation of matrix versus membrane discrimination accuracy.
PEX19-binding Site Validation: The membrane-targeting predictor was validated using peptide-level datasets from human and yeast proteins, including experimentally confirmed PEX19-binding sites and carefully constructed negative controls, with additional validation on phylogenetically distant organisms to assess generalization capability.
External Benchmark Comparison: Performance was compared against DeepLoc-2.0 using their standardized SwissProt benchmark dataset containing 304 peroxisomal proteins and 27,999 non-peroxisomal proteins, ensuring fair comparison with existing general-purpose subcellular localisation tools.
All datasets employed strict redundancy control using UniRef50 clustering and cluster-stratified cross-validation to prevent information leakage and ensure robust performance estimates. External validation sets were completely independent from training data to provide unbiased assessment of generalization capability. The composition of the datasets is summarized in the table below and all the protein sequences used for the benchmarking are available at this link. In parenthesis are reportend the number of unique sequences from which PEX19 binding regions are extracted.
Performance and Accuracy
The is-pero pipeline demonstrates robust performance across all classification tasks through rigorous validation protocols. Each module was evaluated using comprehensive benchmarking strategies to ensure reliable predictions for peroxisomal protein analysis.
Binary Classification (Peroxisomal vs. Non-peroxisomal): The first classification module achieves an AUC of 0.94 ± 0.02 in 10-fold cross-validation with high specificity (TNR = 0.9893 ± 0.0079) and overall accuracy of 0.9844 ± 0.0095. The model demonstrates strong generalization with consistent performance on held-out test data, making it suitable for reliable proteome-wide screening applications.
Sub-compartment Discrimination (Matrix vs. Membrane): The sub-localisation classifier achieves excellent discrimination with AUC = 0.98 on an independent blind test set, representing a substantial improvement over previous implementations. The model maintains high accuracy (0.89) and F1-score (0.88), enabling confident assignment of peroxisomal proteins to their correct sub-compartments.
Membrane-targeting Signal Validation: The PEX19-binding site predictor demonstrates strong performance with AUC = 0.96 ± 0.05 in cross-validation and AUC = 0.93 on external experimental validation. The model achieves accuracy of 0.85 ± 0.05 in cross-validation and maintains reasonable sensitivity (TPR = 0.69) with high specificity (TNR = 0.92) on external validation, effectively distinguishing true membrane-targeting signals.
Comparative Benchmarking: When benchmarked against DeepLoc-2.0 on their standardized dataset, is-pero shows substantial improvements, achieving an MCC of 0.6181 ± 0.0754 compared to 0.5600 ± 0.0800 for DeepLoc-2.0 with ProtT5 embeddings, representing a 10.4% relative improvement in peroxisomal protein prediction accuracy.
All performance metrics were obtained through rigorous validation protocols including nested cross-validation, independent blind test sets, and external experimental validation. The pipeline's performance metrics, particularly its high specificity and AUC values, demonstrate its reliability for both large-scale proteome screening and detailed peroxisomal protein characterization.
References
Please cite the publications below:
Anteghini, M., Martins dos Santos, V.A.P., Saccenti, E. (2021).
In-Pero: Exploiting deep learning embeddings of protein sequences to predict the localisation of peroxisomal proteins
International Journal of Molecular Sciences. 22(12), 6409.
Anteghini, M., Haja, A., Dos Santos, V. A. P. M., Schomaker, L., Saccenti, E. (2023).
OrganelX web server for sub-peroxisomal and sub-mitochondrial protein localisation and peroxisomal
target signal detection. Computational and Structural Biotechnology Journal 21, 128–133.